Über den „großen Haken“ beim Paradepferd der modernen Informatik, den „Künstlichen Neuronalen Netzen“ haben wir schon des öfteren gesprochen: So beeindruckend die Ergebnisse auch sind – wie die Algorithmen letztlich zu ihren Resultaten kommen und was in den internen Schichten genau abläuft, das können selbst die jeweiligen Programmierer nicht genau sagen. In vielen Szenarien reicht der pragmatische Ansatz „Hauptsache, das Ergebnis ist gut“ ja auch; in vielen anderen aber auch nicht. (Wobei eine gewisse sehr problematische Tendenz unverkennbar ist, die Resultate einer vermeintlich objektiven „Künstlichen Intelligenz“ eben nicht zu hinterfragen.)
Aber dieses „Black Box“-Konzept ist ohnehin nicht alternativlos, das zeigen amerikanische Wissenschaftler jetzt im Fachblatt „Nature Methods“.
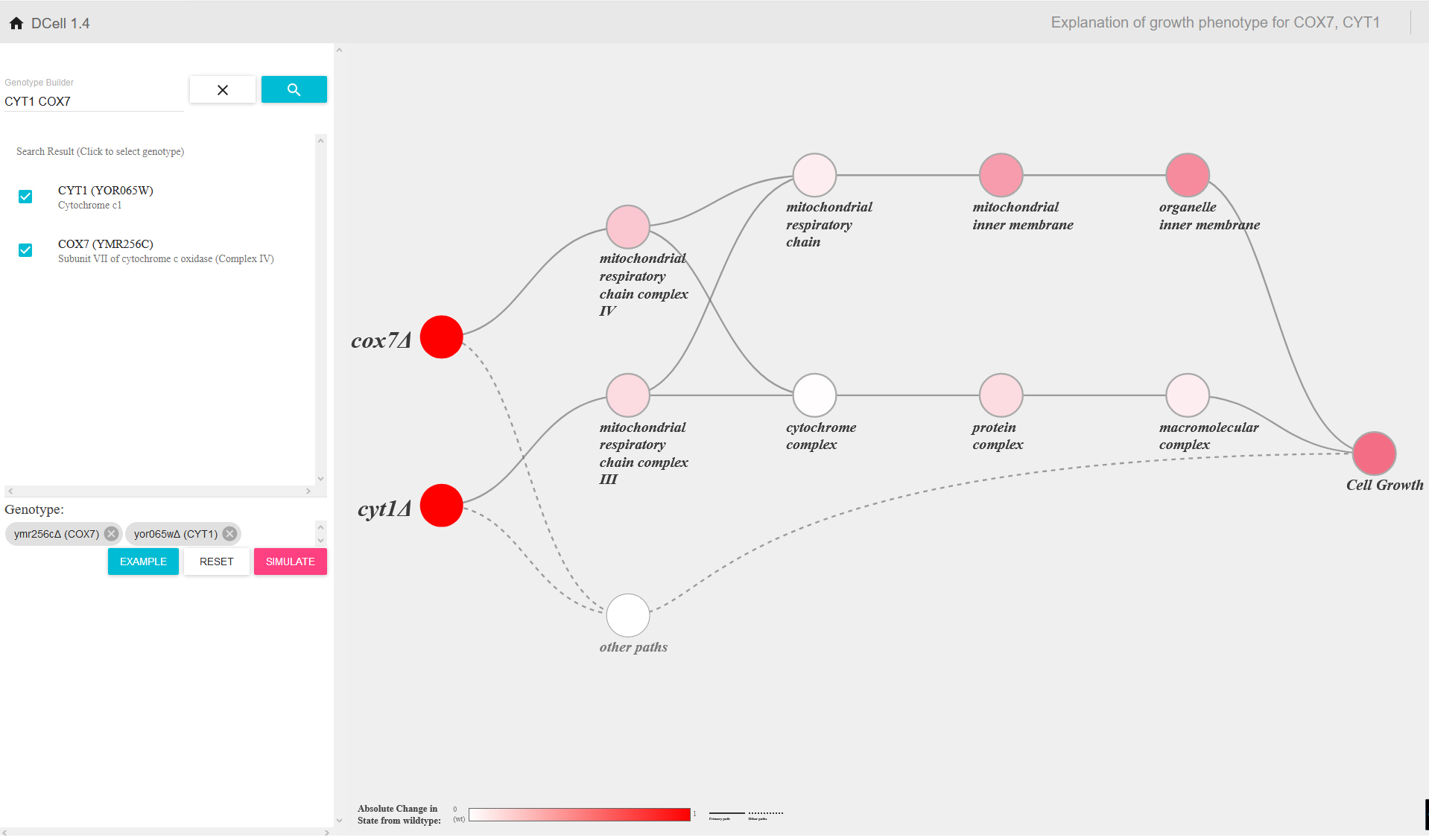
Screenshot from d-cell.ucsd.edu, where researchers can use DCell, a new virtual yeast cell developed at UC San Diego School of Medicine.
Bei „DCell“, dem „Visible neural network“ von Trey Idecker von der University of California San Diego, organisieren sich die internen Schichten („Layer“) und ihre Verknüpfungen im Gegensatz zu herkömmlichen neuronalen Netzen nicht einfach im Training selbst, (als letztlich rein statistisches „Fitting“ zwischen einem „Input“ und einem „Output“-Wert), ihre Architektur ist von vornherein in den wesentlichen Strukturen vorgegeben. Den Bauplan für „DCell“ liefert der bestens erforschte Modellorganismus der Molekularbiologen, die Bierhefe (Saccharomyces cerevisiae).
Die erste, die Eingangsschicht entspricht den einzelnen Genen und ihren Mutationen; und die letzte, die Ausgangsschicht entspricht dem Verhalten der Zelle, nämlich wie schnell sie wächst. Und die Schichten dazwischen, das sind praktisch die physikalischen Größenordnungen in der Zelle, jede folgende Schicht entspricht einer größeren, komplexeren Strukturebene.
Die ersten Schichten des Neuronalen Netzes bilden also den Nanometerbereich ab, wo ein Gen den Bauplan für ein Protein liefert. Die späteren Schichten repräsentieren dann Strukturen wie Membranen, die letzten die größeren Einheiten wie Zellkern oder Mitochondrien. Damit ist das „Visible Neural Network“ im Grunde die digitale Simulation einer kompletten Zelle, die sich nun mit Trainingsdaten füttern lässt. Und die gibt es im Falle der Bierhefe reichlich:
Es gab so viele Studien in den letzten zehn Jahren – wir haben Daten über 12 Millionen einzelne Genmutationen, und da hat jeweils jemand im Labor nachgemessen, welche Auswirkung die Mutation auf das Wachstum der Hefezelle hatte.
Mit dem trainierten Algorithmus konnten Trey Ideker und seine Kollegen anschließend nicht nur die schon bekannten Auswirkungen einzelner Mutationen wie in einem digitalen Modellbaukasten nachvollziehen, sondern sogar in den internen Schichten des Neuralen Netzes noch neue Entdeckungen machen. Es gibt nämlich offenbar selbst bei der intensiv erforschten Bierhefe Proteinkomplexe bzw. „zelluläre Subsysteme“, die bislang unbekannt waren, aber durchaus Auswirkungen auf das Zellwachstum haben. Letztendlich geht es Trey Ideker aber nicht um Bierhefe – er ist Krebsforscher. Er sucht nach Wegen, das Wachstum von Tumorzellen zu verhindern. Und vielleicht könnte eine digitale Zellsimulation auch hier wichtige Erkenntnisse liefern. Was die nötigen Trainingsdaten angeht, ist Ideker recht optimistisch:
Ich schätze mal, so zu Beginn der 2020er Jahre werden wir rund eine Million Krebs-Genome öffentlich verfügbar haben, dann wäre eine Big-Data-Analyse kein Problem. Aber die größere Herausforderung ist: Haben wir genug Wissen über die Biologie von Krebszellen, das wir für die Schichten in unserem Neuronalen Netz brauchen? Nein, haben wir nicht.
Trey Ideker und seine Kollegen, aber auch viele andere Forscher-Teams weltweit arbeiten deshalb intensiv daran, öffentliche Datenbanken über die interne Funktionsweise von Tumorzellen aufzubauen. Mit einer digitalen Zellsimulation, so Idekers Vision, könnte man dann vielleicht auch ein erst in jüngster Zeit erkanntes Problem in den Griff bekommen: Dass nämlich Krebs bei jedem einzelnen Patienten anders funktioniert.
Deswegen brauchen wir dieses Neuronale-Netz-Modell, weil all diese möglichen Varianten in unserem Modell abgebildet wären. Bei dem einen Patienten wird vielleicht die eine Route durch die Ursache-Wirkungs-Hierarchie aktiviert, bei dem zweiten eine andere. Bevor wir so ein komplettes Modell haben, können wir das nicht voraussagen.
Deutschlandfunk – Forschung aktuell vom 06.03.2018 (Moderation: Lennart Pyritz)